505(b)(2)s in context in Europe

You own a medicinal product eligible for the 505(b)(2) pathway in the United States. You also wish to obtain approval in Europe. Now what?
In this article, we go through the routes to marketing authorisation in the US, with emphasis on the 505(b)(2) route. We then identify comparable routes that may be used in the European Economic Area (EEA) to submit 505(b)(2)-type products, indicating the pros and cons of each. Finally, we outline the ways in which Real Regulatory can add value to your strategy setting and submission of your European application.

Routes to marketing authorisation in the US
There are three routes to marketing authorisation in the United States1.
-
- The 505(b)(1) route is a full application (New Drug Application – NDA), supported entirely by the sponsor’s own studies.
- The 505(j) route is the generic route (Abbreviated New Drug Application – ANDA), suitable for off-patent products.
- The 505(b)(2) route lies in-between: applications submitted via this route are NDAs supported partly by the sponsor’s own study(ies) and partly by data not developed by the sponsor (e.g. available in the public domain or pertaining to a reference product developed by another sponsor) rather than generating the full set of data at the sponsor’s own expense.
The 505(b)(2) route has become very popular, constituting 50% of all NDA approvals between 2013 and 20182. Its popularity is due to the reduced development costs whilst allowing differentiation from existing products. Most products approved via the 505(b)(2) pathway are eligible for a number of years of market exclusivity.
Differentiation from the reference product can take many forms, such as a change in dosage form, indication or route of administration; a new combination of active ingredients; a new active ingredient or a new prodrug of an existing drug.
Legal bases comparable to 505(b)(2) in the EEA
In Europe, there are several legal bases for marketing authorisation. These are outlined in Table 1, together with an indication of important characteristics and pros and cons of each route. The table is restricted to small molecule drugs, because there are far fewer choices for biological products
Table 1 Legal bases for marketing authorisation applications in the European Economic Area3
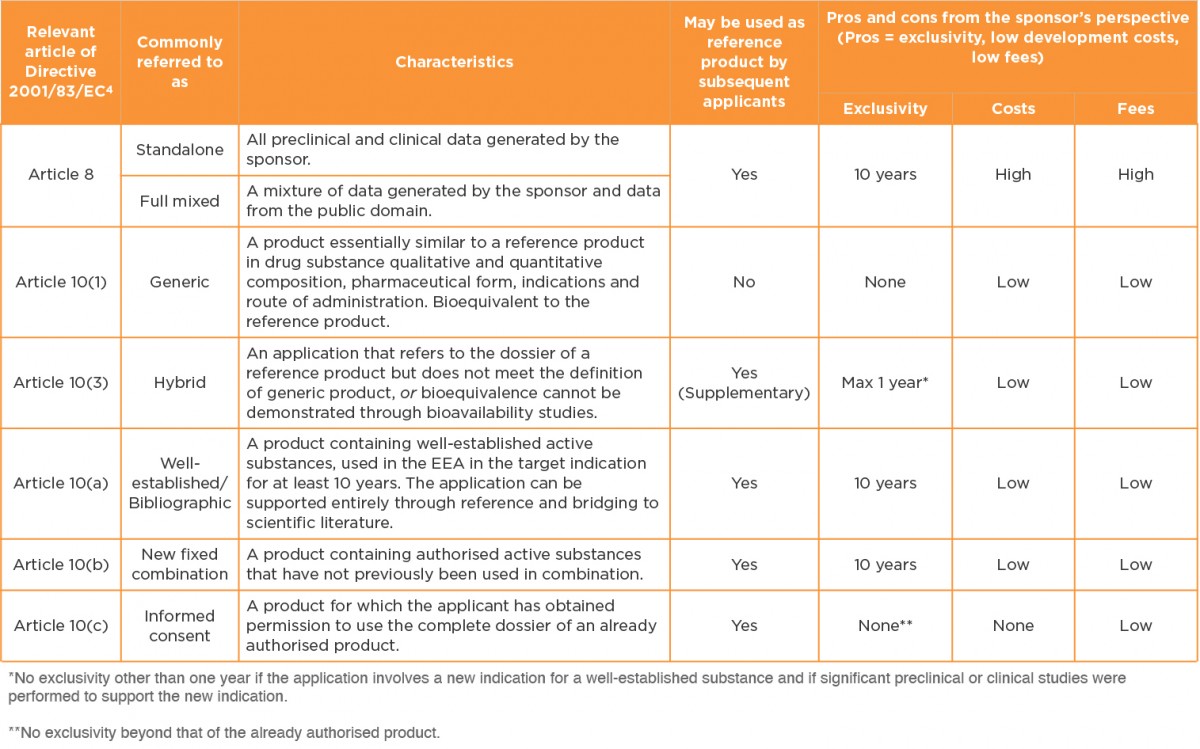
The 505(b)(2) route is often equated with the Article 10(3) hybrid legal basis. However, when one examines the complete list of possible legal bases it is clear that this might not always be the case5.
European legislation restricts the hybrid route to the following cases:
-
- Changes in the active substance(s) (e.g. same therapeutic moiety but different salt form, in cases where proof of similar safety and efficacy is required)
- Change in therapeutic indications
- Change in pharmaceutical form
- Change in strength (quantitative change to the active substance/s)
- Change in route of administration
- Bioequivalence cannot be demonstrated through bioavailability studies (e.g. suprabioavailability or topical products)
In many cases, therefore, applications that are submittable via the 505(b)(2) route in the US can and should be submitted via the Article 10(3) hybrid legal basis in Europe. However, there are plenty of exceptions. Some examples are provided here:


How can we help you?
The European legal basis for your product should ideally be chosen as early as possible, in order to take the requirements of both regions into consideration when designing your development program and obtaining scientific advice. We have all too often seen sponsors having to supplement their US development program with additional studies, most of which could have been avoided if European requirements had been taken into consideration when creating the initial development plan.
Choosing the appropriate legal basis for your marketing authorisation application in the EEA is a complex matter and key to a successful filing. We can help you choose the best pathway for your product depending on your requirements. One size does not fit all and there is a lot to consider, from your target markets and desired positioning to your development budget and aspects dictated by the product itself, such as dosage form and indication.

Furthermore, there are other significant complexities in Europe that we can help you navigate. Here are some of them.
-
- The possibility of saving money by registering as a small to medium-sized enterprise with the European Medicines Agency. You do not have to have an EEA-based company entity to achieve this – you can be annexed to our existing SME status.
- The need to perform clinical trials in children and to obtain approval for a paediatric investigation plan.
- The fragmented nature of scientific advice, which can be obtained centrally from the EMA or from a multitude of national competent authorities. The choice of authorities to approach as well as the timing may be critical.
- Despite the extensive harmonisation between ICH regions, requirements still differ in many respects. We can provide technical consultancy throughout the development process (the earlier the better) to help you achieve a product file which is submittable in Europe.
- Choosing and managing the appropriate regulatory procedure through which to submit the marketing authorisation application for your product: national, decentralised or centralised.
Conclusion
The 505(b)(2) route to marketing authorisation in the United States has become very popular. Companies owning a product registered (or eligible to be registered) via this pathway might also wish to submit their product in Europe. Real Regulatory can help companies navigate the significant complexities involved in choosing an appropriate legal basis for submission, setting regulatory strategy and dealing with scientific advice and marketing authorisation procedures.